- AROMATICITÉ
- AROMATICITÉPeu de notions sont aussi fondamentales et aussi souvent utilisées que celle d’aromaticité; cependant c’est une des moins claires et des plus difficiles à préciser. Elle tire son origine de faits chimiques qui font apparaître d’étroites parentés entre certaines molécules.Ce fut Kekule qui, le premier, reconnut, il y a plus de cent ans, une analogie entre diverses substances extraites d’huiles naturelles: l’aldéhyde benzoïque, le toluène, l’acide benzoïque, l’alcool benzylique auxquels vinrent bientôt s’ajouter le benzène, le phénol, l’aniline, l’acide salicylique et le cymène. Ces molécules possèdent toutes, en effet, un système insaturé contenant six atomes de carbone qui résistent aux transformations et aux dégradations chimiques, à condition qu’elles ne soient pas trop brutales. On peut, par exemple, remplacer des atomes ou des groupements d’atomes portés par ce système, sans l’altérer (réactions de substitution); on peut aussi oxyder ces molécules sans les détruire. D’autre part, étant insaturé, il est capable de fixer trois molécules d’hydrogène ou de chlore, mais ces additions se font sur l’ensemble du système des six atomes en une seule étape, sans qu’il soit possible de mettre en évidence des produits d’addition partielle correspondant à la fixation de une ou deux molécules seulement. Ce fait renforce encore le caractère très particulier de ce sytème de six atomes de carbone, faisant de lui un tout homogène que l’on appelle le système benzénique. Ces mêmes propriétés se rencontrent, à des degrés variables d’ailleurs, dans les carbures à noyaux condensés, comme le naphtalène ou l’anthracène et aussi dans les molécules cycliques contenant des hétéroatomes comme la pyridine, le pyrrole, le furanne ou le thiophène. Mais si dans ces molécules la stabilité et la résistance à l’oxydation sont encore grandes, si des réactions de substitution peuvent encore se produire sans altérer le système insaturé, ce dernier ne forme plus un tout aussi homogène que dans le benzène, car on peut isoler plusieurs produits intermédiaires dans l’hydrogénation ou la fixation d’halogènes, ou réaliser des additions diéniques qui fragmentent le système initial.C’est cet ensemble de propriétés, communes à toutes ces molécules, qui les a fait grouper sous la dénomination d’aromatiques , rappelant ainsi l’origine des premières molécules pour lesquelles ce caractère fut reconnu. Mais il a fallu attendre le développement de la chimie quantique pour arriver à comprendre la raison profonde qui lie ces molécules entre elles.1. Structure électronique des molécules aromatiquesAvant d’aborder le problème de la structure des molécules aromatiques elles-mêmes, il est nécessaire de rappeler quelques résultats généraux obtenus par la mécanique ondulatoire dans l’interprétation de la liaison chimique.Dans un atome, les électrons ne peuvent avoir une énergie arbitraire, mais se placent au contraire sur des niveaux atomiques auxquels correspondent des énergies et des fonctions d’onde d’espace, appelées orbitales atomiques , bien déterminées. L’électron étant aussi caractérisé par une variable de spin qui, par rapport à une direction donnée, ne peut prendre que deux valeurs opposées, sur chaque niveau nous ne pourrons placer que 0, 1 ou 2 électrons, les deux électrons, dans ce dernier cas, correspondant à des valeurs de spin opposées. Enfin le carré du module de la fonction d’onde donne la densité électronique en chaque point. Le problème de la molécule ne diffère de celui de l’atome que par le fait que la charge positive, autour de laquelle gravitent les électrons, n’est pas ponctuelle, mais est répartie sur plusieurs centres: les noyaux des atomes. La description d’une molécule sera nécessairement analogue à celle d’un atome. Les électrons se placeront sur des niveaux moléculaires à raison de 0, 1 ou 2 par niveau. Dans l’état de plus basse énergie, qui est l’état dans lequel se trouvent normalement les molécules à basse température et en l’absence de rayonnement, les électrons se trouveront par paires, de spins opposés, sur les niveaux les plus bas. Si l’énergie correspondant à une telle répartition est inférieure à celle des électrons dans les atomes isolés, l’édifice sera stable: c’est la molécule. La diminution de l’énergie du système quand on passe des atomes isolés à la molécule est l’énergie de liaison.Cette façon de voir la liaison chimique est absolument générale et ne souffre aucune exception. Ce n’est que l’ignorance du phénomène exact qui a fait jadis introduire divers types de liaisons chimiques. La seule différenciation qui apparaît réside dans la répartition spatiale de la densité électronique, c’està-dire, en fait, de la fonction d’onde associée à chaque niveau.Dans le cas d’une molécule comme celle d’hydrogène H2 la fonction d’onde, donc les électrons, est pratiquement entièrement localisée entre les deux noyaux, et la densité électronique est de révolution autour de la ligne des noyaux. Nous écrirons symboliquement H 漣 H, le tiret représentant la paire d’électrons.Le cas de la molécule d’éthylène que l’on écrit symboliquement H2C 略CH2 est plus complexe: chacune des liaisons C 漣 H sera assurée par une paire d’électrons; la liaison entre les deux atomes de carbone se fera par l’intermédiaire de quatre électrons qui, par conséquent, utiliseront deux niveaux moléculaires. Le calcul montre qu’un de ces niveaux correspond à une liaison analogue à celle que l’on trouve dans H2, c’est-à-dire localisée entre les deux noyaux et de révolution, l’autre à un nuage électronique composé de deux parties symétriques situées l’une au-dessus, l’autre au-dessous du plan de la molécule et reliant les atomes de carbone. La fonction d’onde qui décrit ce dernier système est construite sur les orbitales 2pz du carbone. Comme elles, elle possède le plan de la molécule comme plan d’antisymétrie. La densité électronique sera donc symétrique par rapport au plan de la molécule, mais elle sera nulle dans ce plan. Une telle fonction d’onde sera dite de type 神, ainsi, par extension, que la liaison qui lui correspond, par opposition aux autres liaisons (C 漣 H ou la première C 漣 C envisagée) dont la densité électronique est maximale dans le plan de la molécule et que l’on qualifie de 靖. Par un abus de langage, on parlera même d’électrons 靖 et 神. C’est cet ensemble formé d’une liaison 靖 et d’une liaison 神 qui constitue la double liaison , les deux tirets (face=F0019 略) représentant chacun une paire d’électrons comme précédemment, mais étant sous-entendu que les deux paires d’électrons ne sont pas équivalentes. La séparation des électrons en deux classes 靖 et 神 se retrouve chaque fois que la molécule est plane, par exemple dans le butadiène. Toutefois, une situation nouvelle apparaît. Par le même mécanisme que les électrons des orbitales 2pz des atomes de carbone dans l’éthylène formaient une liaison 神 entre ces atomes, l’électron de l’orbitale 2pz de l’atome de carbone 2 pourra se lier à l’électron de l’orbitale 2pz de l’atome de carbone 1 ou 3, si bien que les doubles liaisons ne seront pas indépendantes, mais on aura un nuage électronique 神 réparti sur les quatre atomes de carbone. Au lieu d’être localisés comme dans l’éthylène entre deux atomes, les électrons des doubles liaisons sont délocalisés ; cette situation porte le nom de conjugaison des doubles liaisons.C’est cet état conjugué qui est à l’origine du caractère aromatique. Considérons en effet la molécule de benzène. D’après le schéma classique de Kekule (formule 1), cette molécule devrait posséder trois doubles liaisons. Mais par analogie avec ce que nous avons vu dans le butadiène, les électrons 神 de ces liaisons ne vont pas rester associés par paires localisées pour les doubles liaisons indépendantes, ils vont au contraire participer à un ensemble complètement délocalisé. Dans le butadiène (CH2 略CH 漣CH 略CH2), les liaisons initialement doubles et simples occupent des situations topologiques différentes, malgré la conjugaison, une différence existe encore entre ces deux groupes de liaisons: on sait en effet, par exemple, que leurs longueurs sont différentes (0,134 et 0,147 nm respectivement pour les terminales et la liaison centrale). Dans le benzène, l’équivalence des six groupements CH et de leur position respective impose une symétrie d’ordre 6 pour la molécule. En d’autres termes, la conjugaison se traduira ici par l’uniformisation complète de la densité électronique et par conséquent de toutes les propriétés physico-chimiques des liaisons C 漣 C. En particulier, toutes les liaisons auront la même longueur: 0,14 nm environ.C’est ce que veulent symboliser les formules 2 et 3, formules qui doivent être préférées à celles qui utilisent des simples et des doubles liaisons localisées comme celles de Kekule et qui par là même particularisent les liaisons.La structure des molécules aromatiques condensées généralise celle du benzène. Par exemple, pour le naphtalène (formule 4): le système 神 est formé de dix électrons complètement délocalisés sur l’ensemble des atomes de carbone.Au fur et à mesure que le nombre de cycles benzéniques accolés augmentera, nous aurons un système conjugué de plus en plus étendu, pour atteindre finalement le graphite lorsque tout le plan sera pavé d’hexagones.Il faut cependant remarquer que les molécules aromatiques ne contiennent pas nécessairement des cycles benzéniques. Citons par exemple, parmi les hydrocarbures, l’azulène, qui est un isomère du naphtalène (formule 5). Le système 神 comprend dix électrons répartis sur deux cycles: l’un pentagonal, l’autre heptagonal.De même, une molécule aromatique peut contenir des hétéroatomes. Par exemple, dans la pyridine C5H5N (formule 6), qui dérive formellement du benzène par le remplacement d’un groupement CH par un atome d’azote, c’est ce dernier qui fournit un électron au système conjugué.Il peut arriver aussi que l’hétéroatome apporte deux électrons au système conjugué, dans ce cas le cycle possédera un atome de carbone en moins, par exemple dans le furanne (formule 7), le pyrrole (formule 8) ou le thiophène (formule 9).Ces systèmes peuvent d’ailleurs servir de point de départ pour obtenir d’autres molécules aromatiques, par exemple en leur accolant des cycles benzéniques: indole (formule 10), carbazole (formule 11).Enfin, on notera que la présence d’atomes de carbone n’est pas indispensable, citons par exemple le borazole (formule 12).Le système 神 comprend six électrons qui sont fournis par les atomes d’azote, les atomes de bore n’apportant rien. Cette molécule a des propriétés physiques extrêmement voisines de celles du benzène.2. Propriétés générales des molécules aromatiquesIl résulte de ce qui précède que l’aromaticité est essentiellement liée à la délocalisation des électrons sur toute la molécule. Cette délocalisation entraîne un certain nombre de conséquences.Aspect énergétiqueSi les électrons ne restent pas localisés sur des doubles liaisons indépendantes, mais forment un nuage continu qui s’étend sur toute la molécule, c’est que l’énergie du système est plus basse dans ce dernier état. En d’autres termes, la conjugaison se traduit par une stabilisation du système. L’abaissement de l’énergie est appelé énergie de conjugaison (cette expression est synonyme d’énergie de résonance , mais doit lui être préférée).Le cas du benzène a été particulièrement étudié. La chaleur dégagée par l’hydrogénation d’une double liaison bisubstituée, mais isolée, est de 112 kJ par mole. Si les trois doubles liaisons étaient indépendantes comme le veut la formule de Kekule, la chaleur d’hydrogénation de la molécule de benzène serait de 336 kJ. Or, en réalité, elle n’est que de 208 kJ. C’est que l’énergie de la molécule réelle conjuguée est inférieure de 150 kJ à l’énergie de la formule hypothétique de Kekule. On obtient le même résultat en considérant les chaleurs de combustion.On notera que l’hexatriène:
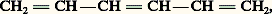 qui contient trois doubles liaisons conjuguées, forme un système beaucoup moins stable et non aromatique: son énergie de conjugaison est seulement de quelques kJ/mole. Cet exemple nous montre donc l’importance d’avoir une conjugaison cyclique.À titre indicatif, on peut donner les énergies de conjugaison de quelques molécules aromatiques, énergies calculées à partir de la formule de Kekule:DIR\benzène150 kJnaphtalène322 kJanthracène485 kJazulène192 kJ/DIRPlus l’énergie de conjugaison est grande, plus le système est stable. Comme application, nous signalerons le cas de l’azulène et du naphtalène, qui sont isomères et dont les formules de Kekule de référence ont même énergie puisqu’elles contiennent le même nombre de doubles et de simples liaisons. L’azulène est donc moins stable que le naphtalène, ce qui explique qu’une élévation de température en présence de catalyseur (soufre) le transforme en naphtalène.Incidence sur la géométrie moléculaireNous avons vu que dans le benzène la délocalisation des électrons entraînait l’uniformisation des longueurs de liaisons. Ce phénomène est absolument général, mais l’uniformisation ne peut être aussi parfaite dans toutes les molécules où les liaisons occupent des positions respectives différentes. Par exemple, dans le naphtalène, les distances interatomiques sont (en nm):
qui contient trois doubles liaisons conjuguées, forme un système beaucoup moins stable et non aromatique: son énergie de conjugaison est seulement de quelques kJ/mole. Cet exemple nous montre donc l’importance d’avoir une conjugaison cyclique.À titre indicatif, on peut donner les énergies de conjugaison de quelques molécules aromatiques, énergies calculées à partir de la formule de Kekule:DIR\benzène150 kJnaphtalène322 kJanthracène485 kJazulène192 kJ/DIRPlus l’énergie de conjugaison est grande, plus le système est stable. Comme application, nous signalerons le cas de l’azulène et du naphtalène, qui sont isomères et dont les formules de Kekule de référence ont même énergie puisqu’elles contiennent le même nombre de doubles et de simples liaisons. L’azulène est donc moins stable que le naphtalène, ce qui explique qu’une élévation de température en présence de catalyseur (soufre) le transforme en naphtalène.Incidence sur la géométrie moléculaireNous avons vu que dans le benzène la délocalisation des électrons entraînait l’uniformisation des longueurs de liaisons. Ce phénomène est absolument général, mais l’uniformisation ne peut être aussi parfaite dans toutes les molécules où les liaisons occupent des positions respectives différentes. Par exemple, dans le naphtalène, les distances interatomiques sont (en nm):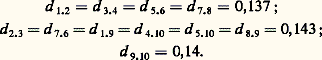 Dans l’azulène, les liaisons périphériques mesurent toutes approximativement 0,14 nm, mais la liaison binucléaire 0,147 nm. Dans le graphite, toutes les distances C 漣 C sont évidemment égales: 0,142 nm.Charges électroniquesOn peut penser a priori que la délocalisation des électrons va se traduire par un déplacement de charge électronique. Initialement neutres, les atomes vont se charger positivement ou négativement selon qu’ils vont s’appauvrir ou s’enrichir en électrons 神. Dans une molécule comme le benzène, la symétrie de la molécule interdit un tel déplacement, mais il n’en sera pas de même dans toutes les molécules.Considérons d’abord les hydrocarbures. Deux cas se présentent: ou bien la molécule ne contient que des cycles pairs (hydrocarbures benzénoïdes), ou bien elle possède au moins un cycle impair (fulvène, azulène et molécules dérivées: benzofulvène, benzoazulène...). Dans les premières molécules, appelées encore hydrocarbures alternants , les charges nettes des atomes de carbone sont pratiquement nulles, si bien que le moment dipolaire de ces molécules est très faible, quelques dixièmes de debye au plus (à moins que par raison de symétrie celui-ci soit identiquement nul). Dans les secondes, qualifiées de non alternantes , les charges nettes peuvent être assez importantes et la molécule est polaire. Par exemple, pour le fulvène le moment est de l’ordre de un debye, pour l’azulène il est aussi de cet ordre de grandeur; pour des molécules plus volumineuses, il peut atteindre plusieurs debyes.Le cas des molécules qui contiennent des hétéroatomes est un peu différent. Ici encore plusieurs cas sont à distinguer. Si la molécule ne contient qu’un hétéroatome plus électronégatif que l’atome de carbone, cas qui se rencontre dans la pyridine, l’atome d’azote sera porteur d’une charge négative. Mais si l’hétéroatome participe à la conjugaison par deux électrons (pyrrole), nécessairement il s’appauvrira en électrons 神, l’écoulement sera évidemment d’autant plus faible que l’atome sera plus électronégatif. Dans tous les cas, les molécules seront polaires.Spectres d’absorptionDans une molécule comme dans un atome, l’absorption d’une radiation de fréquence 益 est possible si l’énergie qui lui correspond h 益 (h = C te de Planck) est exactement égale à la différence d’énergie entre un état excité et l’état fondamental. Les fréquences d’absorption caractérisent donc un système. Par exemple, les doubles liaisons C=C bisubstituées absorbent vers 180 nm.La délocalisation des électrons entraîne un remaniement complet des niveaux électroniques, si bien que les doubles liaisons ne seront pas décelables par absorption. Par exemple, le benzène absorbera dès 250 nm. Sans entrer dans le détail de l’interprétation des spectres des molécules aromatiques, nous dirons que lorsque le système conjugué s’agrandit, si la molécule résultante est une molécule alternante , la première bande d’absorption se déplace vers les grandes longueurs d’onde (déplacement bathochrome ). Si la molécule n’est pas alternante, le déplacement peut être soit bathochrome, soit se faire en sens inverse: déplacement hypsochrome. Par exemple:DIR\benzène255 nmnaphtalène275 nmanthracène370 nmpentacène580 nm/DIRSpectres infrarouge et RamanLes fréquences qui apparaissent dans le spectre d’absorption infrarouge ou de diffusion Raman sont égales à certaines fréquences de vibration des molécules. Comme les fréquences d’absorption électroniques, elles caractérisent les molécules ou même, la plupart du temps, certains groupes d’atomes: doubles liaisons C=C, groupement carbonyle... La conjugaison, qui se traduit par une délocalisation des électrons, modifie ces fréquences. Une délocalisation complète, comme celle qui se produit dans les molécules aromatiques, provoque l’apparition de fréquences très caractéristiques propres à chaque système: système benzénique, système naphtalénique, etc., si bien qu’à la seule vue de ces spectres on peut identifier le système aromatique de la molécule.Aspect chimiqueNous avons vu que la conjugaison cyclique entraîne à la fois une modification des liaisons et une stabilisation générale de la molécule. Ce sont ces deux faits qui vont conditionner la réactivité chimique des molécules aromatiques. Toutes ces molécules possèdent des électrons 神, énergétiquement moins liés que les électrons 靖, ce seront eux qui réagiront. Ils seront attaqués essentiellement par des systèmes avides d’électrons (réactifs électrophiles): ions positifs ou radicaux. Le principal type de réaction susceptible de se produire est la substitution électrophile. Prenons comme exemple le benzène. Soit R+ un édifice positif (ions NO+2, S3H+, Cl+ ou R+) produit dans le milieu par une réaction préliminaire. Cet ion attaquera un sommet et se liera au carbone. Pour se former, la liaison C 漣 R devra utiliser deux électrons qui seront soustraits au système conjugué. Le système benzénique est alors détruit. Le système conjugué ne comprend plus que 4 électrons répartis sur 5 atomes de carbone (formule 13). Un tel système est instable et, pour reconstituer son cycle benzénique, la molécule expulsera soit l’ion R+, soit l’ion H+. Dans le premier cas, la réaction redonnera le benzène; dans le second, un benzène substitué C6H5R. On voit donc que le cycle benzénique a été reconstitué pour des raisons énergétiques.Dans le cas du benzène, tous les sommets sont équivalents, mais il n’en est pas de même dans d’autres hydrocarbures, comme l’anthracène ou l’azulène. Le sommet attaqué sera celui sur lequel il sera le plus facile d’amener deux électrons pour réaliser la liaison avec l’ion arrivant: position 見 (ou 1, 4, 5, 8) dans le naphtalène, sommet 1 dans l’azulène. Mais, dans tous les cas, le mécanisme reste le même.À côté des réactions de substitution qui laissent le système conjugué intact, il y a les réactions qui le détruisent, comme les réactions d’addition. Si la molécule est symétrique (cas du benzène), on obtient d’emblée la saturation du système conjugué: fixation de trois molécules d’hydrogène, de chlore ou d’ozone. Mais, dans les autres cas, les additions peuvent être moins poussées. Ce critère sera utile pour évaluer le degré d’aromaticité. Par exemple, l’hydrogénation du naphtalène par le sodium dans l’ammoniac liquide conduit au 1,4-dihydronaphtalène (formule 14), qui se transforme d’ailleurs en 1,2-dihydronaphtalène plus stable. La bromation du phénanthrène conduit au dérivé dibromé en -9,10 (formule 15).De même les hétérocycles donnent des fixations réduisant le système conjugué sans le détruire totalement. Signalons par exemple des réactions diéniques sur le furanne (fixation d’acide acétylène dicarboxylique; réaction 16) ou sur le pyrrole (polymérisation).Cette dernière se produit plus difficilement. En effet, l’atome d’azote étant moins électronégatif que l’atome d’oxygène, il retient moins les électrons, donc la délocalisation est plus grande et par conséquent l’uniformisation des propriétés des liaisons rend moins probables de telles réactions. L’effet est encore plus net dans le thiophène où de telles réactions n’ont plus lieu, alors que les réactions de substitution sont plus faciles que dans le furanne ou le pyrrole.La présence de substituants dans les molécules aromatiques apporte évidemment des modifications parfois importantes à la structure de la molécule, surtout si le substituant est susceptible de se conjuguer avec le système aromatique. C’est le cas du styrolène ou, plus encore, de l’aldéhyde ou de l’acide benzoïque.La conjugaison avec un atome porteur d’un doublet est plus faible, car les électrons de celui-ci sont retenus par l’électronégativité de l’atome porteur du doublet plus grande que celle du carbone (ex. chlorobenzène, phénol, aniline), si bien que le système aromatique conserve ses caractéristiques générales.Un dernier exemple de réactions intéressantes à citer est relatif aux molécules aromatiques substituées par des chaînes carbonées saturées. En général, elles sont attaquées, de préférence au système aromatique, à cause de la stabilité de ce dernier. La plus importante de ces réactions est l’oxydation qui coupe les chaînes latérales, laissant à leur place le groupement C2H. Une oxydation plus ménagée conduit à des aldéhydes ou à des cétones:
Dans l’azulène, les liaisons périphériques mesurent toutes approximativement 0,14 nm, mais la liaison binucléaire 0,147 nm. Dans le graphite, toutes les distances C 漣 C sont évidemment égales: 0,142 nm.Charges électroniquesOn peut penser a priori que la délocalisation des électrons va se traduire par un déplacement de charge électronique. Initialement neutres, les atomes vont se charger positivement ou négativement selon qu’ils vont s’appauvrir ou s’enrichir en électrons 神. Dans une molécule comme le benzène, la symétrie de la molécule interdit un tel déplacement, mais il n’en sera pas de même dans toutes les molécules.Considérons d’abord les hydrocarbures. Deux cas se présentent: ou bien la molécule ne contient que des cycles pairs (hydrocarbures benzénoïdes), ou bien elle possède au moins un cycle impair (fulvène, azulène et molécules dérivées: benzofulvène, benzoazulène...). Dans les premières molécules, appelées encore hydrocarbures alternants , les charges nettes des atomes de carbone sont pratiquement nulles, si bien que le moment dipolaire de ces molécules est très faible, quelques dixièmes de debye au plus (à moins que par raison de symétrie celui-ci soit identiquement nul). Dans les secondes, qualifiées de non alternantes , les charges nettes peuvent être assez importantes et la molécule est polaire. Par exemple, pour le fulvène le moment est de l’ordre de un debye, pour l’azulène il est aussi de cet ordre de grandeur; pour des molécules plus volumineuses, il peut atteindre plusieurs debyes.Le cas des molécules qui contiennent des hétéroatomes est un peu différent. Ici encore plusieurs cas sont à distinguer. Si la molécule ne contient qu’un hétéroatome plus électronégatif que l’atome de carbone, cas qui se rencontre dans la pyridine, l’atome d’azote sera porteur d’une charge négative. Mais si l’hétéroatome participe à la conjugaison par deux électrons (pyrrole), nécessairement il s’appauvrira en électrons 神, l’écoulement sera évidemment d’autant plus faible que l’atome sera plus électronégatif. Dans tous les cas, les molécules seront polaires.Spectres d’absorptionDans une molécule comme dans un atome, l’absorption d’une radiation de fréquence 益 est possible si l’énergie qui lui correspond h 益 (h = C te de Planck) est exactement égale à la différence d’énergie entre un état excité et l’état fondamental. Les fréquences d’absorption caractérisent donc un système. Par exemple, les doubles liaisons C=C bisubstituées absorbent vers 180 nm.La délocalisation des électrons entraîne un remaniement complet des niveaux électroniques, si bien que les doubles liaisons ne seront pas décelables par absorption. Par exemple, le benzène absorbera dès 250 nm. Sans entrer dans le détail de l’interprétation des spectres des molécules aromatiques, nous dirons que lorsque le système conjugué s’agrandit, si la molécule résultante est une molécule alternante , la première bande d’absorption se déplace vers les grandes longueurs d’onde (déplacement bathochrome ). Si la molécule n’est pas alternante, le déplacement peut être soit bathochrome, soit se faire en sens inverse: déplacement hypsochrome. Par exemple:DIR\benzène255 nmnaphtalène275 nmanthracène370 nmpentacène580 nm/DIRSpectres infrarouge et RamanLes fréquences qui apparaissent dans le spectre d’absorption infrarouge ou de diffusion Raman sont égales à certaines fréquences de vibration des molécules. Comme les fréquences d’absorption électroniques, elles caractérisent les molécules ou même, la plupart du temps, certains groupes d’atomes: doubles liaisons C=C, groupement carbonyle... La conjugaison, qui se traduit par une délocalisation des électrons, modifie ces fréquences. Une délocalisation complète, comme celle qui se produit dans les molécules aromatiques, provoque l’apparition de fréquences très caractéristiques propres à chaque système: système benzénique, système naphtalénique, etc., si bien qu’à la seule vue de ces spectres on peut identifier le système aromatique de la molécule.Aspect chimiqueNous avons vu que la conjugaison cyclique entraîne à la fois une modification des liaisons et une stabilisation générale de la molécule. Ce sont ces deux faits qui vont conditionner la réactivité chimique des molécules aromatiques. Toutes ces molécules possèdent des électrons 神, énergétiquement moins liés que les électrons 靖, ce seront eux qui réagiront. Ils seront attaqués essentiellement par des systèmes avides d’électrons (réactifs électrophiles): ions positifs ou radicaux. Le principal type de réaction susceptible de se produire est la substitution électrophile. Prenons comme exemple le benzène. Soit R+ un édifice positif (ions NO+2, S3H+, Cl+ ou R+) produit dans le milieu par une réaction préliminaire. Cet ion attaquera un sommet et se liera au carbone. Pour se former, la liaison C 漣 R devra utiliser deux électrons qui seront soustraits au système conjugué. Le système benzénique est alors détruit. Le système conjugué ne comprend plus que 4 électrons répartis sur 5 atomes de carbone (formule 13). Un tel système est instable et, pour reconstituer son cycle benzénique, la molécule expulsera soit l’ion R+, soit l’ion H+. Dans le premier cas, la réaction redonnera le benzène; dans le second, un benzène substitué C6H5R. On voit donc que le cycle benzénique a été reconstitué pour des raisons énergétiques.Dans le cas du benzène, tous les sommets sont équivalents, mais il n’en est pas de même dans d’autres hydrocarbures, comme l’anthracène ou l’azulène. Le sommet attaqué sera celui sur lequel il sera le plus facile d’amener deux électrons pour réaliser la liaison avec l’ion arrivant: position 見 (ou 1, 4, 5, 8) dans le naphtalène, sommet 1 dans l’azulène. Mais, dans tous les cas, le mécanisme reste le même.À côté des réactions de substitution qui laissent le système conjugué intact, il y a les réactions qui le détruisent, comme les réactions d’addition. Si la molécule est symétrique (cas du benzène), on obtient d’emblée la saturation du système conjugué: fixation de trois molécules d’hydrogène, de chlore ou d’ozone. Mais, dans les autres cas, les additions peuvent être moins poussées. Ce critère sera utile pour évaluer le degré d’aromaticité. Par exemple, l’hydrogénation du naphtalène par le sodium dans l’ammoniac liquide conduit au 1,4-dihydronaphtalène (formule 14), qui se transforme d’ailleurs en 1,2-dihydronaphtalène plus stable. La bromation du phénanthrène conduit au dérivé dibromé en -9,10 (formule 15).De même les hétérocycles donnent des fixations réduisant le système conjugué sans le détruire totalement. Signalons par exemple des réactions diéniques sur le furanne (fixation d’acide acétylène dicarboxylique; réaction 16) ou sur le pyrrole (polymérisation).Cette dernière se produit plus difficilement. En effet, l’atome d’azote étant moins électronégatif que l’atome d’oxygène, il retient moins les électrons, donc la délocalisation est plus grande et par conséquent l’uniformisation des propriétés des liaisons rend moins probables de telles réactions. L’effet est encore plus net dans le thiophène où de telles réactions n’ont plus lieu, alors que les réactions de substitution sont plus faciles que dans le furanne ou le pyrrole.La présence de substituants dans les molécules aromatiques apporte évidemment des modifications parfois importantes à la structure de la molécule, surtout si le substituant est susceptible de se conjuguer avec le système aromatique. C’est le cas du styrolène ou, plus encore, de l’aldéhyde ou de l’acide benzoïque.La conjugaison avec un atome porteur d’un doublet est plus faible, car les électrons de celui-ci sont retenus par l’électronégativité de l’atome porteur du doublet plus grande que celle du carbone (ex. chlorobenzène, phénol, aniline), si bien que le système aromatique conserve ses caractéristiques générales.Un dernier exemple de réactions intéressantes à citer est relatif aux molécules aromatiques substituées par des chaînes carbonées saturées. En général, elles sont attaquées, de préférence au système aromatique, à cause de la stabilité de ce dernier. La plus importante de ces réactions est l’oxydation qui coupe les chaînes latérales, laissant à leur place le groupement C2H. Une oxydation plus ménagée conduit à des aldéhydes ou à des cétones: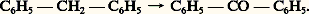 Citons aussi les réactions de substitution qui se font sur le groupement méthyle du toluène sous l’action de la lumière:
Citons aussi les réactions de substitution qui se font sur le groupement méthyle du toluène sous l’action de la lumière: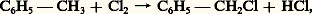 sans altérer le système benzénique.3. Définitions de l’aromaticitéDans les divers exemples étudiés, le caractère aromatique commun à toutes ces molécules se manifestait à des degrés divers. Dans le benzène, il atteignait son maximum d’intensité, mais dans les autres molécules benzéniques, il était quelque peu atténué, dans les hétérocycles on observait une diminution progressive de l’aromaticité quand on passait du thiophène au pyrrole et au furanne. Il serait donc intéressant de trouver un critère qui permette de savoir non seulement si une molécule est aromatique ou non mais aussi, éventuellement, à quel degré elle l’est.La première tentative fondée sur la structure électronique est due à Hückel qui énonça la règle suivante: «Une molécule est aromatique si le nombre d’électrons 神 qu’elle contient est de la forme 4 n + 2, n étant un entier positif.» Le benzène, tous les carbures benzéniques (naphtalène, anthracène), l’azulène satisfont à cette règle, de même les hétérocycles comme la pyridine, le furanne, le pyrrole ou le thiophène. Cette règle lui permit de prévoir l’aromaticité des ions C5H 漣5 et C7H+7 avant même leur découverte. Malheureusement, cette règle rencontre des difficultés, par exemple pour le pyrène (formule 17) que l’ensemble des propriétés indique comme aromatique et qui possède 16 électrons. Pour résoudre la difficulté, on peut modifier l’énoncé de la règle en ne considérant que les électrons portés par les atomes périphériques (soit 14, ici), responsables des réactions de substitution. Malgré cela, toutes les difficultés ne sont pas réglées, puisque le carbure aromatique représenté par la formule 18 ne satisfait pas à la règle, même modifiée.On peut aussi songer à s’adresser à l’énergie de conjugaison pour chiffrer l’aromaticité, puisque la stabilité de ces molécules est une des manifestations les plus frappantes de l’aromaticité. Mais des difficultés apparaissent: en effet, l’énergie de conjugaison du naphtalène est supérieure à celle du benzène, bien qu’il soit moins aromatique. Il en est de même lorsqu’on considère le couple azulène-naphtalène.Craig a proposé une règle fondée sur la symétrie de la molécule. Elle ne s’applique qu’à des molécules qui possèdent un plan de symétrie. Pour savoir si une molécule est aromatique, on écrit sa formule de Kekule, on marque chaque extrémité des doubles liaisons des signes 見 et 廓 de façon que l’on ait le moins de signes de même espèce au contact. Soit f le nombre de paires d’atomes qui s’échangent dans une symétrie par rapport au plan de symétrie, et soit g le nombre de couples de symboles 見 et 廓 qui s’échangent dans la symétrie.Pentalène (formule 19)Pyrène (formule 20)Mais des difficultés apparaissent lorsque l’on peut écrire plusieurs formes de Kekule. Par exemple pour l’hydrocarbure de Hafner:Selon la formule 21Selon la formule 22On élude le problème en disant qu’une telle molécule est pseudo-aromatique. Mais de toute façon la règle de Craig, pas plus que celle de Hückel, ne peut distinguer le degré d’aromaticité dans les hydrocarbures pour lesquels la somme f + g est paire comme dans les benzéniques où pourtant des différences sensibles apparaissent. La raison de leur échec réside dans le fait qu’elles se sont éloignées de l’idée que suggère l’expérience: l’aromaticité est liée à une uniformisation des propriétés des liaisons, nous ajouterons même des liaisons périphériques qui sont les responsables de la réactivité. Comme critère, nous pouvons par exemple nous adresser à la géométrie des molécules. Plus les liaisons auront des longueurs voisines, plus la molécule sera aromatique. D’autre part, l’aromaticité est liée à la délocalisation des électrons, donc à leur facilité de circulation dans la molécule. L’aromaticité pourra être mesurée par la grandeur qui tient compte de ces deux facteurs:
sans altérer le système benzénique.3. Définitions de l’aromaticitéDans les divers exemples étudiés, le caractère aromatique commun à toutes ces molécules se manifestait à des degrés divers. Dans le benzène, il atteignait son maximum d’intensité, mais dans les autres molécules benzéniques, il était quelque peu atténué, dans les hétérocycles on observait une diminution progressive de l’aromaticité quand on passait du thiophène au pyrrole et au furanne. Il serait donc intéressant de trouver un critère qui permette de savoir non seulement si une molécule est aromatique ou non mais aussi, éventuellement, à quel degré elle l’est.La première tentative fondée sur la structure électronique est due à Hückel qui énonça la règle suivante: «Une molécule est aromatique si le nombre d’électrons 神 qu’elle contient est de la forme 4 n + 2, n étant un entier positif.» Le benzène, tous les carbures benzéniques (naphtalène, anthracène), l’azulène satisfont à cette règle, de même les hétérocycles comme la pyridine, le furanne, le pyrrole ou le thiophène. Cette règle lui permit de prévoir l’aromaticité des ions C5H 漣5 et C7H+7 avant même leur découverte. Malheureusement, cette règle rencontre des difficultés, par exemple pour le pyrène (formule 17) que l’ensemble des propriétés indique comme aromatique et qui possède 16 électrons. Pour résoudre la difficulté, on peut modifier l’énoncé de la règle en ne considérant que les électrons portés par les atomes périphériques (soit 14, ici), responsables des réactions de substitution. Malgré cela, toutes les difficultés ne sont pas réglées, puisque le carbure aromatique représenté par la formule 18 ne satisfait pas à la règle, même modifiée.On peut aussi songer à s’adresser à l’énergie de conjugaison pour chiffrer l’aromaticité, puisque la stabilité de ces molécules est une des manifestations les plus frappantes de l’aromaticité. Mais des difficultés apparaissent: en effet, l’énergie de conjugaison du naphtalène est supérieure à celle du benzène, bien qu’il soit moins aromatique. Il en est de même lorsqu’on considère le couple azulène-naphtalène.Craig a proposé une règle fondée sur la symétrie de la molécule. Elle ne s’applique qu’à des molécules qui possèdent un plan de symétrie. Pour savoir si une molécule est aromatique, on écrit sa formule de Kekule, on marque chaque extrémité des doubles liaisons des signes 見 et 廓 de façon que l’on ait le moins de signes de même espèce au contact. Soit f le nombre de paires d’atomes qui s’échangent dans une symétrie par rapport au plan de symétrie, et soit g le nombre de couples de symboles 見 et 廓 qui s’échangent dans la symétrie.Pentalène (formule 19)Pyrène (formule 20)Mais des difficultés apparaissent lorsque l’on peut écrire plusieurs formes de Kekule. Par exemple pour l’hydrocarbure de Hafner:Selon la formule 21Selon la formule 22On élude le problème en disant qu’une telle molécule est pseudo-aromatique. Mais de toute façon la règle de Craig, pas plus que celle de Hückel, ne peut distinguer le degré d’aromaticité dans les hydrocarbures pour lesquels la somme f + g est paire comme dans les benzéniques où pourtant des différences sensibles apparaissent. La raison de leur échec réside dans le fait qu’elles se sont éloignées de l’idée que suggère l’expérience: l’aromaticité est liée à une uniformisation des propriétés des liaisons, nous ajouterons même des liaisons périphériques qui sont les responsables de la réactivité. Comme critère, nous pouvons par exemple nous adresser à la géométrie des molécules. Plus les liaisons auront des longueurs voisines, plus la molécule sera aromatique. D’autre part, l’aromaticité est liée à la délocalisation des électrons, donc à leur facilité de circulation dans la molécule. L’aromaticité pourra être mesurée par la grandeur qui tient compte de ces deux facteurs: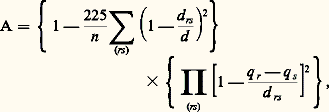 n étant le nombre de liaisons périphériques (rs ) de longueur d rs , d leur longueur moyenne, q r la charge de l’atome r (la constante 225 a été choisie de façon que l’aromaticité de la formule de Kekule soit nulle).Voici quelques résultats:
n étant le nombre de liaisons périphériques (rs ) de longueur d rs , d leur longueur moyenne, q r la charge de l’atome r (la constante 225 a été choisie de façon que l’aromaticité de la formule de Kekule soit nulle).Voici quelques résultats:
● aromaticité nom féminin (latin médiéval aromaticitas) Ensemble de propriétés physiques et chimiques caractéristiques des hydrocarbures aromatiques et de leurs dérivés.⇒AROMATICITÉ, subst. fém.Qualité de ce qui est aromatique (Lar. 19e, LITTRÉ, Nouv. Lar. ill.).Rem. Noté ds QUILLET 1965 avec la mention ,,rare``.PRONONC. — Seule transcription ds LITTRÉ : a-ro-ma-ti-si-té.ÉTYMOL. ET HIST. — 1. Ca 1390 « odeur spécifique d'une chose » (CONTY, Probl. d'Aristote, Bibl. nat., 210, f° 72 r° ds GDF. Compl. : Ceste odeur ou aromaticité), attest. isolée; 2. 2e moitié XIVe s. « qualité de ce qui est aromatique » (Jardin de Santé, I, 31, ibid. : Le romarin a vertus de conforter par son aromaticité) — av. 1590, Paré ds HUG.; répertorié ds la lexicogr. dep. Lar. 19e.Empr. au lat. médiév. aromaticitas au sens 1, XIIe s. URSO, Gloss., 12, p. 37, 13 ds Mittellat. W. s.v., 972, 54; au sens 2, av. 1161 MATTHAEUS PLATEARIUS, Gloss., p. 367, ibid., 972, 62.
Encyclopédie Universelle. 2012.